AS保守程度的统计
甲基化程度的检测
将多重校验的结果做成bed文件
awk '{print $1,$2,$2,$6}' OFS="\t" fdr.file >fdr.bed制作基因组的bed文件
Ghir_A11 48563862 48571977 Ghir_A11G021610
Ghir_A11 48086239 48087160 Ghir_A11G021550
Ghir_A11 48315032 48323201 Ghir_A11G021570使用intersectBed取交集
~/software/bedtools2-2.29.0/bin/intersectBed -loj -a ../TM-1_gene.bed -b CpG_context_TM1_qs_rep1_count.txt_binom.txt_fdr.bed >3计算甲基化程度
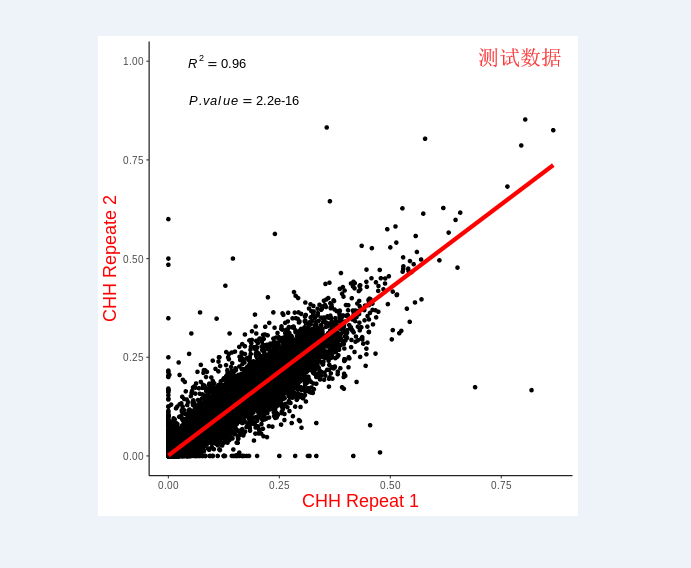
检测重复性
计算pearson相关系数
画图
参考
Last updated