Pan-Genome数据比对
使用bwa进行序列比对
1.构建索引
bwa index /data/cotton/Unmapped_Gb_Mate/Gbarbadense_genome.fasta
bwa index /public/home/cotton/public_data/PAN_Final/GbPAN/Gb.final.1k.fa2.进行比对
bwa mem -t 5 索引前称 sample_1.fq sample_2.fq -o out.bam3.格式转换
4.基因组位置和PAN-genom位置映射
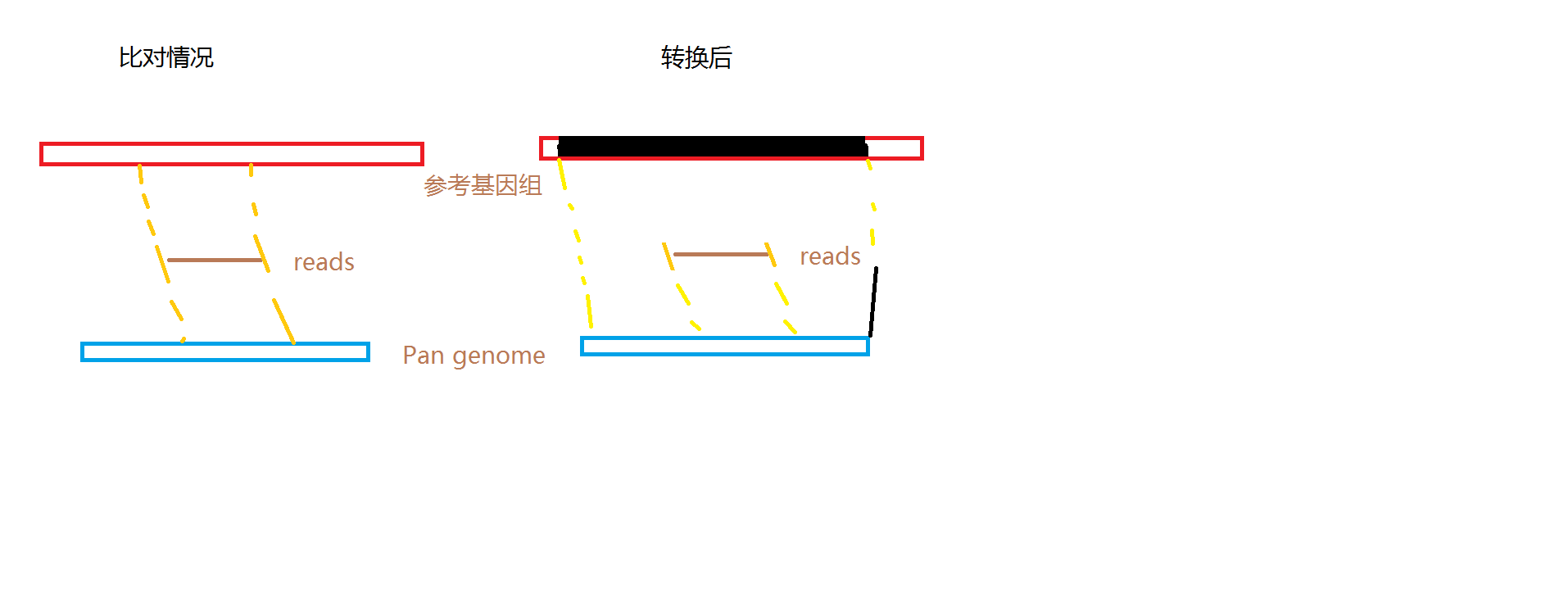
与gene和promoter取交集
使用到的脚本
Last updated